Next-generation sequencing technologies have allowed for sequencing at a low cost and fast speed, and is used more and more to study microbial communities. RNA-seq metatranscriptome and WGS metagenome studies aim to investigate microbial communities at genome and transcriptome levels. In this article, I will introduce a few tools that I frequently use to analyze metagenomic and metatranscriptomic datasets.
Generating Microbial community taxonomy profiles
Since a variety of microbes live in the microbial community at differential relative abundances, the first question researchers usually ask is who is present and at what relative abundance. Kraken (https://ccb.jhu.edu/software/kraken2/) is one of the most frequently used tools to classify microbial community taxonomic information. Kraken uses a K-mer based searching algorithm to assign taxonomic labels to the reads (Fig. 1). Another frequently used tool is MetaPhlan (http://huttenhower.sph.harvard.edu/metaphlan2), which uses clade-specific marker genes to study the microbiome taxonomic composition (Fig. 2). Both approaches are popularly used in microbiome researches and efficient in run time.
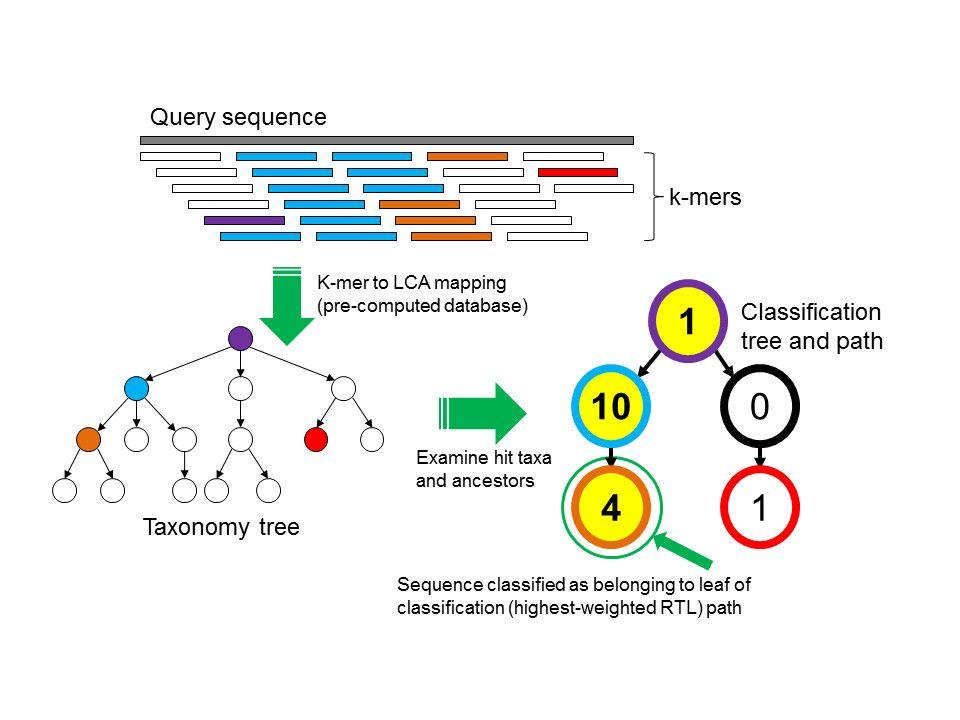
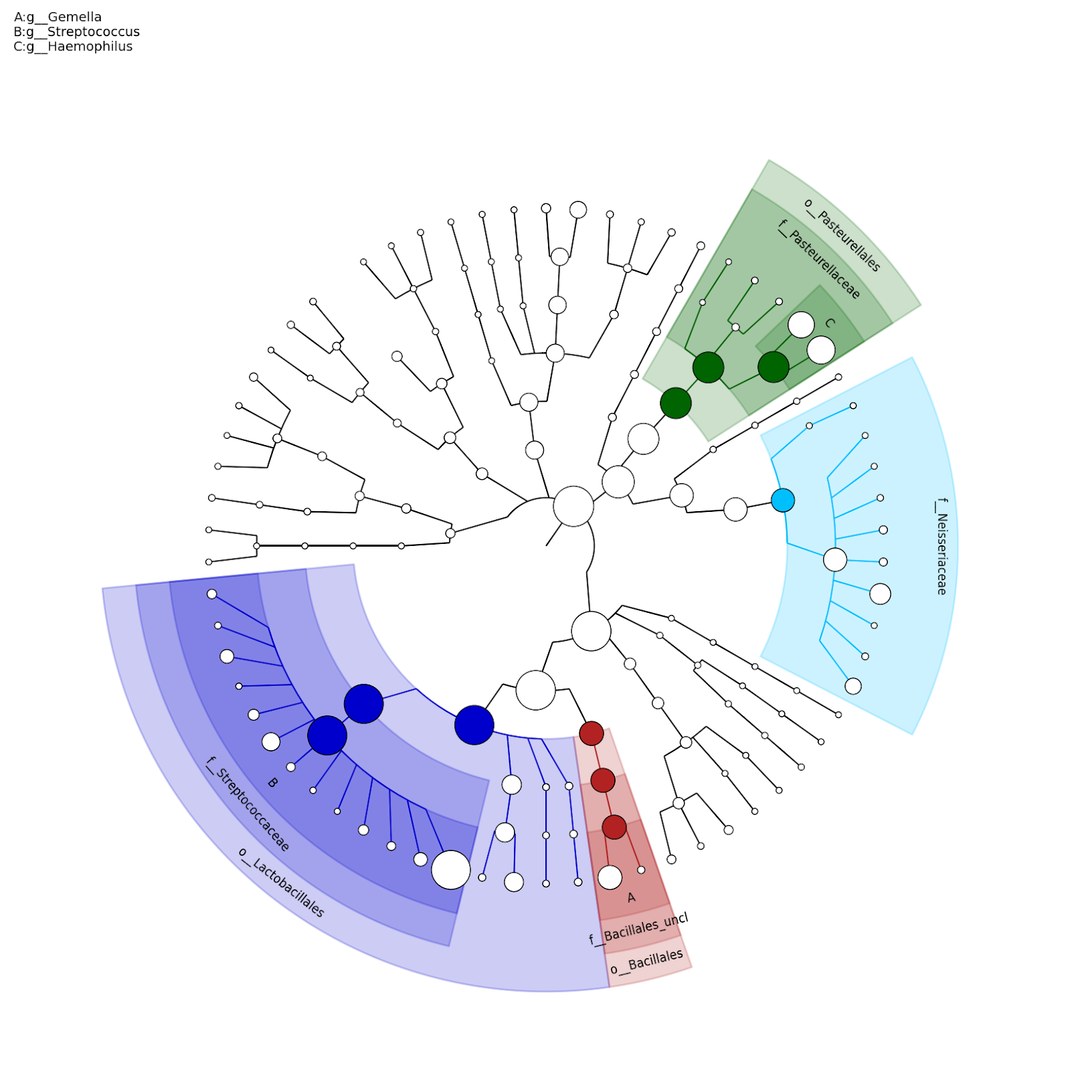
Figures from Wood and Salzberg, 2014 and https://bitbucket.org/nsegata/metaphlan/wiki/MetaPhlAn_Pipelines_Tutorial
Generating Microbial community gene expression profiles
Another question researchers investigate with regards to metagenomic and metatranscriptomic datasets is the presence/expression of bacterial genes in the microbial community. HUMAnN2 is one of the most popular tools in analyzing the bacterial gene expression profiles. Different levels of information can be learned through running HUMAnN2, the reads are first assigned to bacterial taxa and both the mapped and unmapped reads are searched against the protein databases for gene assignments. Gene family abundance, pathway abundance and coverage can be learned from the HUMAnN2 output.
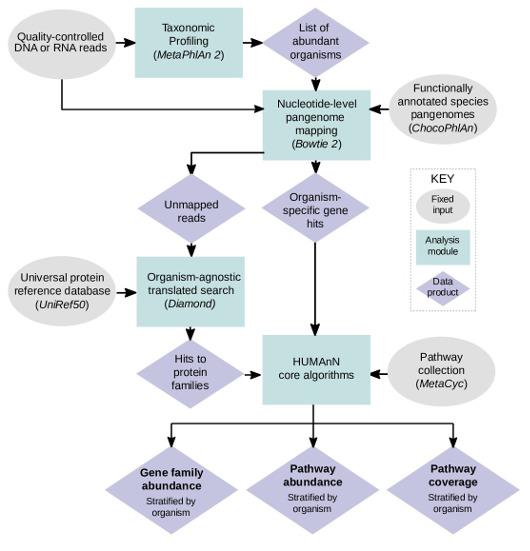
Figure from https://bitbucket.org/biobakery/humann2/wiki/Home
Metagenome and metatranscriptome assembly
A different approach to investigate the microbial community is through assembling the reads into contigs. This allows researchers to identify novel bacterial genomes and genes. A lot of assemblers are designed specifically for metagenomic datasets, such as metaSPAdes and MEGAHIT. These assemblers use k-mer based De Bruijn graphs, which have advantages in handling errors in reads and DNA repeats and used a lot in metagenome assembly.
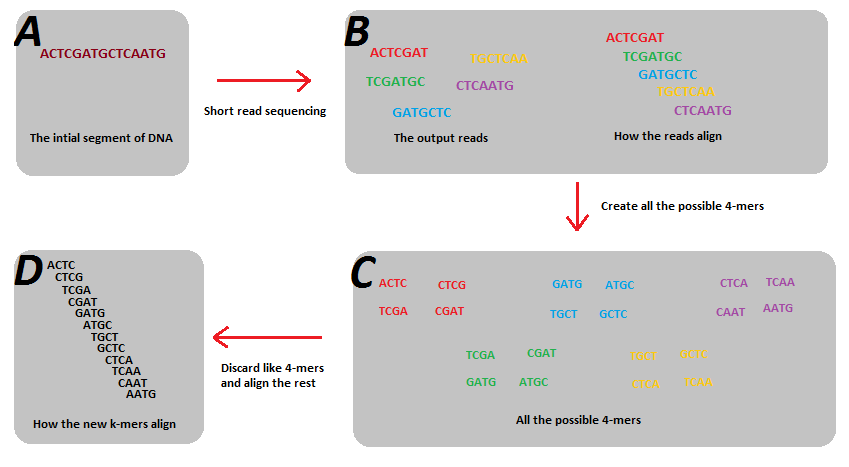
Figure from https://en.wikipedia.org/wiki/K-mer
Summary
Metagenomic and metatranscriptomic datasets contain vast amounts of information including taxonomy classification and gene expression information. Many tools have been developed to extract information from these datasets to allow researchers investigate the microbial community and ask questions of their interest. I am only briefly introducing a few popular tools developed for different purposes. I hope this can be helpful for people who are interested in microbiome research and looking for software to learn the information they are interested in. For more details about the above-mentioned software please see the links and the references below.
For a step-by-step tutorial on analyzing metagenomic data using the above mentioned tools, check out the tutorial at: https://learn.gencore.bio.nyu.edu/metgenomics/
Additional material
1. Nurk, S., et al., metaSPAdes: a new versatile metagenomic assembler. Genome Res, 2017. 27(5): p. 824-834.
2. Franzosa, E.A., et al., Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods, 2018. 15(11): p. 962-968.
3. Li, D., et al., MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics, 2015. 31(10): p. 1674-6.


2 Comments
Mohammed Khalfan · 2019-12-11 at 11:58 pm
Metagenomics research is changing the way we think and live. The tools described here (Kraken, MetaPhlan, HUMAnN2, metaSPAdes, MEGAHIT) are some of the tools driving these changes. For a user-friendly web app which uses another popular software tool called QIIME, check out http://dnasubway.org (the purple line).
Laura Korobkova · 2022-01-20 at 7:43 pm
Hi! Thanks for the guide! Can you please clarify if Kraken can be used as metatranscriptome tool or only metagenome tool? Thank you!