Update 7/21/2021: reform has officially been published as an NFT. Read about this experiment in scientific publishing here. Access the reform publication (PDF) here.
Update 11/20/2019: reform is now available as a web app https://reform.bio.nyu.edu/
reform is a python-based command line tool that allows for fast, easy and robust editing of reference genome sequence and annotation files. With the increase in use of genome editing tools such as CRISPR/Cas9, and the use of reference genome based analyses, the ability to edit existing reference genome sequences and annotations to include novel sequences and features (e.g. transgenes, markers) is increasingly necessary. reform provides a fast, easy, reliable, and reproducible solution for creating modified reference genome data for use with standard analysis pipelines and the 10x Cell Ranger pipeline.
The Problem
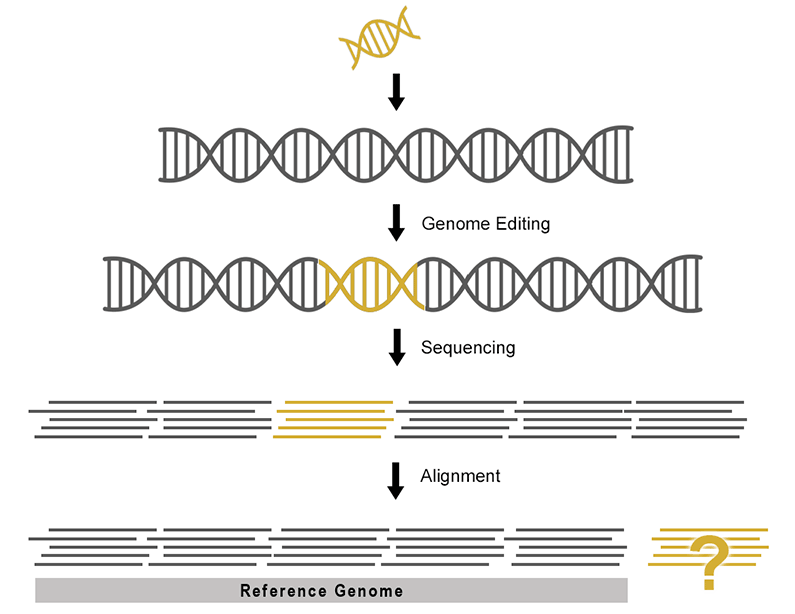
While it is possible to edit reference sequences and annotations manually, the process is time consuming and prone to human error. Data that appears correct to the eye after manual editing may break strict file formatting rules rendering the data unusable during analysis.
The Solution
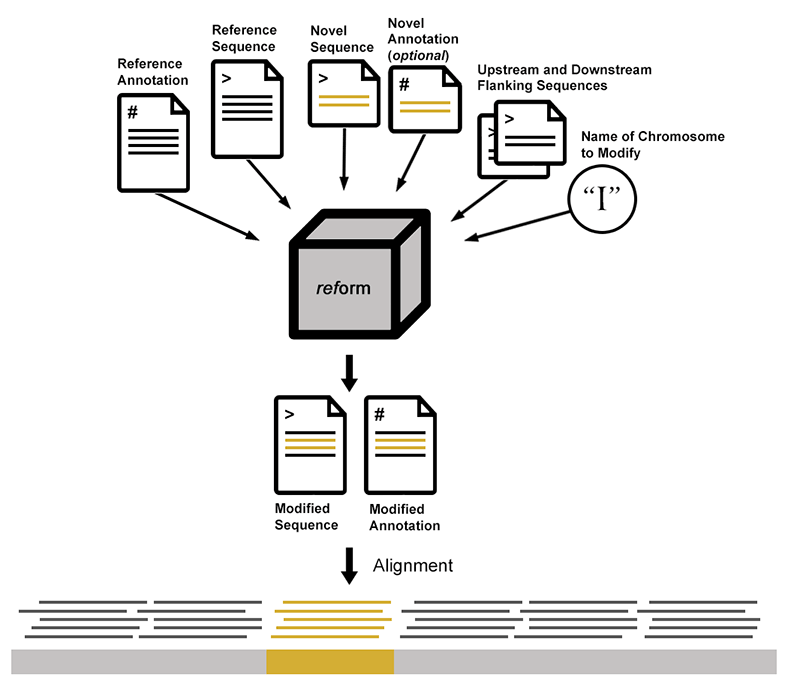
Execution of reform requires a reference sequence (fasta), reference annotation (GFF or GTF), the novel sequences to be added (fasta), and corresponding novel annotations (GFF or GTF). A user provides as arguments the name of the modified chromosome and either the position at which the novel sequence is inserted, or the upstream and downstream sequences flanking the novel sequences. This results in the addition and/or deletion of sequence from the reference in the modified fasta file. In addition to the novel annotations, any changes to the reference annotations that result from deleted or interrupted sequence are incorporated into the modified gff. Importantly, modified gff and fasta files include a record of the modifications.
The position at which the novel sequence is to be inserted can be provided in two different ways:
1) Provide position at which to insert novel sequence.
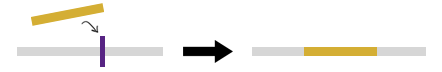
2) Provide upstream and downstream flanking sequences, resulting in deletion of reference sequence between these two regions and addition of the novel sequence.

Download
https://github.com/gencorefacility/reform
Usage
reform requires Python3 and Biopython v1.78 or higher.
On the Greene HPC at NYU, simply load the required module:
module load reform/1
Invoke the python script like this:
reform.py \ --chrom=<chrom> \ --position=<pos> \ --in_fasta=<in_fasta> \ --in_gff=<in_gff> \ --ref_fasta=<ref_fasta> \ --ref_gff=<ref_gff>
Parameters
chrom ID of the chromsome to modify
position Position in chromosome at which to insert <in_fasta>. Can use -1 to add to end of chromosome. Note: Either position, or upstream AND downstream sequence must be provided.
upstream_fasta Path to Fasta file with upstream sequence. Note: Either position, or upstream AND downstream sequence must be provided.
downstream_fasta Path to Fasta file with downstream sequence. Note: Either position, or upstream AND downstream sequence must be provided.
in_fasta Path to new sequence to be inserted into reference genome in fasta format.
in_gff Path to GFF file describing new fasta sequence to be inserted.
ref_fasta Path to reference fasta file.
ref_gff Path to reference gff file.
Example
reform.py \
--chrom="I" \
--upstream_fasta="data/up.fa" \
--downstream_fasta="data/down.fa" \
--in_fasta="data/new.fa" \
--in_gff="data/new.gff" \
--ref_fasta="data/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa" \
--ref_gff="data/Saccharomyces_cerevisiae.R64-1-1.34.gff3"Output
reformed.fa Modified fasta file.
reformed.gff3 Modified GFF file.
An Example Using 10x Cell Ranger
If you’re using the Cell Ranger pipeline, you’ll need to modify your GTF file with reform and then run cellranger makeref to create the new genome data needed for cellranger count. Here’s an example:
1) Prepare reference data using reform
We’ll need the original reference in fasta format, and a fasta file containing the novel sequence to be incorporated into the reference sequence. We’ll also need the original reference GTF file, and a GTF file with the annotations corresponding to the novel sequence.
Let’s get a reference sequence:
greene:/scratch/work/cgsb/genomes/Public/Fungi/Saccharomyces_cerevisiae/Ensembl/R64-1-1/Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa
And the reference GTF:
greene:/scratch/work/cgsb/genomes/genomes/Public/Fungi/Saccharomyces_cerevisiae/Ensembl/R64-1-1/Saccharomyces_cerevisiae.R64-1-1.34.gtf
Here’s the novel sequence we’re adding to the reference (new.fa)
>new_gene TGAAATTCATAGCTGTAGAAAATAAAGAAGCACTCATTTGGTCCATATCCAAACTCATTT AGTGGTTATGCTGGGTATCGTAAACAAAAATGGAGCCAAATGCAGTTATTAATCGTTTAT TGGAGGATCGCAAATTACTACGCTCATCTTTTGTTGGAGAACTACCAATTGCTGCAGTGA CAACGCATATAGCAGTGATTTGTTTCCAAATGAAAGCTGCTGTCTTTGCGTCGTCGATAA
Here’s the GTF corresponding to the novel sequence in new.fa (new.gtf):
I reform exon 1 240 . + 0 gene_id "new_gene"; transcript_id "new_gene.1"; I reform CDS 1 238 . + 0 gene_id "new_gene"; transcript_id "new_gene.1"; I reform start_codon 1 3 . + 0 gene_id "new_gene"; transcript_id "new_gene.1"; I reform stop_codon 238 240 . + 0 gene_id "new_gene"; transcript_id "new_gene.1";
In this example, we’ll add this new gene at position 2650 on chromosome “I”:
reform.py \ --chrom="I" \ --position=2650 \ --in_fasta="new.fa" \ --in_gff="new.gff" \ --ref_fasta="Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa" \ --ref_gff="Saccharomyces_cerevisiae.R64-1-1.34.gtf"
We should have two new files reformed.fa and reformed.gtf which we’ll need in the next step.
2) Run Cell Ranger makeref
cellranger mkref \ --genome=output_genome \ --fasta=reformed.fa \ --genes=reformed.gtf
This will produce a directory called output_genome which we’ll need to provide for the next step
3) Run Cell Ranger count
cellranger count \ --id=run_1 \ --transcriptome=output_genome \ --fastqs=/path/to/fastq_path/ \ --sample=sample_1
4) Putting it together
Here’s an sbatch script that runs everything together and makes use of resources on the HPC
#!/bin/sh
#
#SBATCH --verbose
#SBATCH --job-name=reform_ranger
#SBATCH --output=reform_ranger_%j.out
#SBATCH --error=reform_ranger_%j.err
#SBATCH --time=12:00:00
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=20
#SBATCH --mem=110000
#SBATCH --mail-user=${USER}@nyu.edu
module load cellranger/2.1.0
module load reform/1
reform.py \
--chrom="I" \
--position=2650 \
--in_fasta="new.fa" \
--in_gff="new.gff" \
--ref_fasta="Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa" \
--ref_gff="Saccharomyces_cerevisiae.R64-1-1.34.gff3"
cellranger mkref \
--genome=output_genome \
--fasta=reformed.fa \
--genes=reformed.gtf \
--nthreads=20 \
--memgb=100
cellranger count \
--id=run_1 \
--transcriptome=output_genome \
--fastqs=/path/to/fastq_path/ \
--sample=sample_1


6 Comments
Sally · 2018-10-16 at 5:57 pm
Works like a charm, thanks!
Anthony · 2023-02-15 at 2:24 pm
Is it possible to modify multiple sequences with reform?
Mohammed Khalfan · 2023-02-15 at 3:31 pm
At the moment reform only supports one modification at a time.
Nestor Saiz · 2023-05-09 at 6:58 pm
I keep getting the following error when running from shell:
reform.py: error: the following arguments are required: –in_fasta, –in_gff, –ref_fasta, –ref_gff
even though I provide all arguments as described here. When I try to submit via the web app, I am told both upstream and downstream FASTA files are required, even though I’m providing both. Please assist!
Elisabet · 2023-07-05 at 10:35 am
Great tool! Is it possible to substitute rows for new ones in the GFF? When adding a new sequence, the annotations of all downstream genes should be changed, could I use reform for this purpose?
Priscila · 2023-12-06 at 1:39 pm
Is it possible to add the new sequence to the reference and it’s respective gtf, but to an “artificial” chromosome (chromosome not present in the reference)?