In the Kenneth Birnbaum Lab, we are interested in understanding how the plant root is able to grow continuously over the plant’s life and maintain its specific root structure (Fig.1). More specifically, what is the role of the stem cells present at the very tip of the root (QC and initials), in this maintenance and development.

The fascinating part of plant growth in general is its capacity of continuous growth over the plant’s life. This is due to a very specific set of embryonic cells located at the very tip of the root or shoot, respectively called the Root or Shoot Apical meristem (RAM or SAM). In the case of the root, these cells slowly divide to give rise to the different cell types constituting the root. From these first divisions, cells continue to divide and differentiate, forming a gradient until they fully mature to form the functional root.
During this process very specific sets of genes are successively activated and repressed to allow this slow maturation along the meristem. In addition, these distinct genetic programs are different depending on the cell type composing the meristem. However, so far technical limitations have prevented researchers from having full knowledge of these specific genes.
The power of single cell RNA seq (scRNA-seq) allows us to get a complete snapshot of every cell composing the meristem and recreate a transcriptome map allowing us to decipher the multiple mechanisms of cell maturation.
At the Genomics Core at NYU, we use Illumina sequencing and the new Chromium system from 10X Genomics to analyze data.
This new procedure of single cell RNA-seq is actually quite simple:
The first crucial step is to get the single cells from the root tip of our plant of interest. To perform this step, we rely on a cocktail of enzymes able to digest the complex cell wall matrix that binds the cells together. We do this as follows:
Seedlings of the plant model Arabidopsis thaliana are grown in vitro on MS medium, and at the appropriate age root tips are sectioned and transferred into our cocktail of enzymes. After 1-2h the cell wall is digested, and cells are freely released in solution. Cells are then washed from the enzyme, carefully counted to assess their quality and viability and then directly processed through the 10X chromium system.
This machine combines together, through a microfluidic device, the plant cells with poly-T beads and releases them into a droplet of oil. The final product is an emulsion of thousands of droplets containing the cells and the beads (Fig.2).

The cDNA synthesis is performed directly inside each droplet allowing for rapid and easy processing, and preventing RNA degradation. As each bead contains a distinct specific barcode, cDNA from the same droplet get the same barcode necessary to sequence and identify the cell.
Sequencing of the 10x libraries is performed on site by the Genomics Core team and data is aligned on the genome of interest using software provided by 10x genomics called Cell Ranger. Data are then analyzed using the R package Seurat, developed at NYU and the New York Genome Center by the team of Rahul Satija.
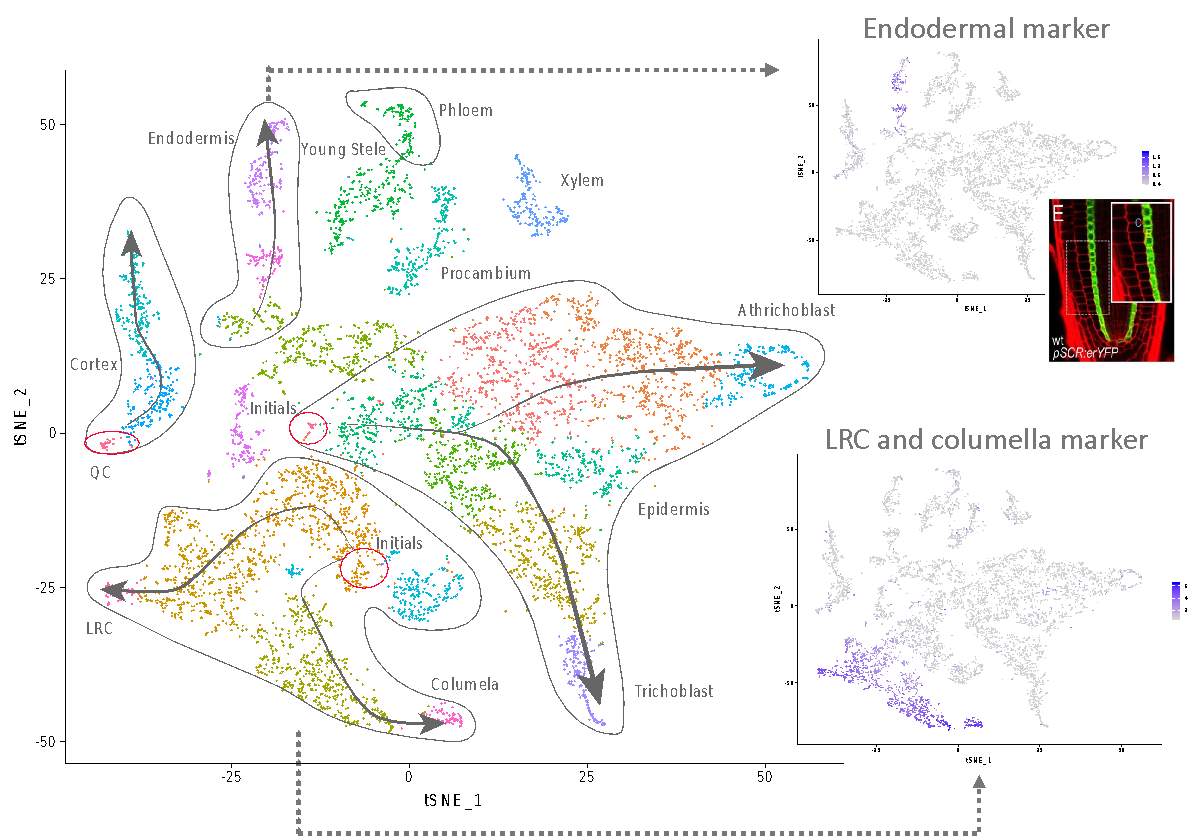
Seurat allows for the clustering of all sequenced cells depending on their respective transcriptome. Identification of cluster cell identity is performed by comparing marker gene expression and previously published transcriptome data (Fig. 3).
Results obtained allow us to draw a map containing every cell type present in the meristem of the plant model Arabidopsis thaliana. In addition, we are able to draw the maturation path of the cells by identifying younger and older tissue (Fig. 3 arrows). This map is of unprecedented accuracy and constitutes the starting point to identify new genes and mechanisms specific to each cell type.
Moreover, single cell analysis can be performed on Arabidopsis mutants, meristem after wounding, or specific treatments to identify cell type specific response and genetic programs involved. This is the ongoing work performed in Kenneth Birnbaum lab, and specifically on root regeneration.


1 Comment
Mohammed Khalfan · 2019-12-12 at 12:03 am
This is a great article. The last paragraph is particularly exciting. Looking forward to seeing what’s coming.