This is an updated version of the variant calling pipeline post published in 2016 (link). This updated version employs GATK4 and is available as a containerized Nextflow script on GitHub.
Identifying genomic variants, including single nucleotide polymorphisms (SNPs) and DNA insertions and deletions (indels), from next generation sequencing data is an important part of scientific discovery. At the NYU Center for Genomics and Systems Biology (CGSB) this task is central to many research programs. For example, the Carlton Lab analyzes SNP data to study population genetics of the malaria parasites Plasmodium falciparum and Plasmodium vivax. The Gresham Lab uses SNP and indel data to study adaptive evolution in yeast, and the Lupoli Lab in the Department of Chemistry uses variant analysis to study antibiotic resistance in E. coli.
To facilitate this research, a bioinformatics pipeline has been developed to enable researchers to accurately and rapidly identify, and annotate, sequence variants. The pipeline employs the Genome Analysis Toolkit 4 (GATK4) to perform variant calling and is based on the best practices for variant discovery analysis outlined by the Broad Institute. Once SNPs have been identified, SnpEff is used to annotate, and predict, variant effects.
This pipeline is intended for calling variants in samples that are clonal – i.e. a single individual. The frequencies of variants in these samples are expected to be 1 (for haploids or homozygous diploids) or 0.5 (for heterozygous diploids). To call variants in samples that are heterogeneous, such as human tumors and mixed microbial populations, in which allele frequencies vary continuously between 0 and 1 researcher should use GATK4 Mutect2 which is designed to identify subclonal events (workflow coming soon).
Base Quality Score Recalibration (BQSR) is an important step for accurate variant detection that aims to minimize the effect of technical variation on base quality scores (measured as Phred scores). As with the original pipeline (link), this pipeline assumes that a ‘gold standard’ set of SNPS and indels are not available for BQSR. In the absence of a gold standard the pipeline performs an initial step detecting variants without performing BQSR, and then uses the identified SNPs as input for BQSR before calling variants again. This process is referred to as bootstrapping and is the procedure recommended by the Broad Institute’s best practices for variant discovery analysis when a gold standard is not available.
This pipeline uses nextflow, a framework that enables reproducible and portable workflows (https://www.nextflow.io/). The full pipeline and instructions on how to use the Nextflow script are described below.
Script Location
greene
/scratch/work/cgsb/scripts/variant_calling/gatk4/main.nf- You don’t need to copy the script, but you should copy the file
nextflow.configfrom the above directory into a new project directory.
(/scratch/work/cgsb/scripts/variant_calling/gatk4/nextflow.config) - If you want to copy and run the
main.nfscript locally, you must copy the/bindirectory as well. This needs to be in the same directory asmain.nf. - You will set parameters for your analysis in the
nextflow.configfile - See How to Use and Examples below to learn how to configure and run the script.
- This Nextflow script uses pre-installed software modules on the NYU HPC, and is tailored to the unique file naming scheme used by the Genomics Core at the NYU CGSB.
github
https://github.com/gencorefacility/variant-calling-pipeline-gatk4
- This version of the script is configured for standard Illumina filenames
- This version of the script does not use software modules local to the NYU HPC, it is instead packaged with a Docker container which has all tools used in the pipeline. Integration with Docker allows for completely self-contained and truly reproducible analysis. This example shows how to run the workflow using the container. The container is hosted on Docker at: https://hub.docker.com/r/gencorefacility/variant-calling-pipeline-gatk4
Full List of Tools Used in this Pipeline
- GATK4
- BWA
- Picard Tools
- Samtools
- SnpEff
- R (dependency for some GATK steps)
How to Use This Script
Setting Up Nextflow
This pipeline is written in Nextflow, a computational pipeline framework. The easiest way to get setup with Nextflow is with conda. On the NYU HPC, conda is already installed and available as a module.
module load anaconda3/2019.10
Note: Use module avail anaconda to check for the latest conda version.
Update: NYU HPC users can load Nextflow using the module load command. There is no need to install via conda. Simply load the module and skip to “Setting Up The Script” below.
For users outside NYU, see the conda installation instructions, then come back and follow the rest of the instructions below.
Once you have conda loaded, make sure you add the bioconda channel (required for Nextflow) as well as the other channels bioconda depends on. It is important to add them in this order so that the priority is set correctly (if you get warnings that a channel exists and is being moved to the top, that’s OK keep going):
conda config --add channels defaults
conda config --add channels bioconda
conda config --add channels conda-forge
Create a conda environment for Nextflow. I call it ‘nf-env’ but you can call it anything you like.
conda create --name nf-env
(If you get a message asking you to update conda, press enter to proceed and allow your conda environment to be created.)
We’ll install Nextflow inside this newly created environment. Anytime we want to use Nextflow, we’ll need to load the environment first using the following command:
conda activate nf-env
When a conda environment has been loaded, you’ll see its name in parenthesis at the command prompt, for example:
(nf-env) [user@log-1 ~]$
Once you’ve activated your conda environment, install nextflow using:
conda install nextflow
(When you’re finished with nextflow, deactivate the environment using conda deactivate)
Setting Up The Script
Once you have Nextflow setup, we can run the pipeline. The pipeline requires six mandatory input parameters to run. The values can be hard coded into a config file, or provided as command line arguments (examples below).
1) params.reads The directory with your FastQ libraries to be processed, including a glob path matcher. Must include a paired end group pattern matcher (i.e. {1,2}).
Example:params.reads = "/path/to/data/*_n0{1,2}_*.fastq.gz"
or params.reads = "/path/to/data/*_R{1,2}_*.fastq.gz"
2) params.ref Reference genome to use, in fasta format. If you’re at the NYU CGSB, there’s a good chance your genome of interest is located in the Shared Genome Resource (/scratch/work/cgsb/genomes/Public/). Selecting a reference genome from within the Shared Genome Resource ensures that all index files and reference dictionaries required by BWA, Picard, GATK, etc. are available. If you’re using your own reference data, you will need to build index files for BWA (using BWA), a fasta index (using samtools), and a reference dictionary (using Picard Tools). These files need to be located in the same directory as the reference fasta file.
3) params.snpeff_dbThe name of the SnpEff database to use. To determine the name of the SnpEff DB to use, ensure you have snpeff installed/loaded and then issue the databases command. # On the NYU HPC you can load SnpEff using module load
module load snpeff/4.3i
# $SNPEFF_JAR is the path to the snpEff.jar file.
# On the NYU HPC this is automatically set to the
# snpEff.jar path when the module is loaded
java -jar $SNPEFF_JAR databases | grep -i SPECIES_NAME
From the list of returned results, select the database you wish to use. The value of the first column is the value to input for params.snpeff_db.
For example if you wanted to use the Arabidopsis Thaliana SnpEff database:
java -jar $SNPEFF_JAR databases | grep -i thaliana
Output:
athalianaTair10 Arabidopsis_Thaliana http://downloads.sourceforge.net/...
Then:
params.snpeff_db='athalianaTair10'
Note: If your organism is not available in SnpEff, it is possible to create a custom SnpEff Database if a reference fasta and gff file are available.
4) params.outdir By default, output files will go into params.outdir/out, temporary GATK files will go into params.outdir/gatk_temp, SnpEff database files will go into params.outdir/snpeff_db, and Nextflow will run all analysis in params.outdir/nextflow_work_dir.These individual paths can be overridden by specifying the params.out, params.tmpdir, params.snpeff_data, and the workDir parameters respectively. Note: There is no params. prefix for workDir.
5) params.pl Platform. E.g. Illumina (required for read groups)
6) params.pm Machine. E.g. nextseq (required for read groups)
Examples
Note: If you are working on the NYU HPC, you should run all nextflow jobs in an interactive slurm session. This is because Nextflow uses some resources in managing your workflow.
Once you have set up your nextflow environment you can perform your analysis in multiple ways. Below I describe four different ways this can be done.
Scenario 1: Hard code all parameters in config file
Copy the nextflow.config file into a new directory (see Script Location). Edit the first six parameters in this file, e.g.:
params.reads = "/path/to/reads/*_n0{1,2}_*.fastq.gz"
params.ref = "/path/to/ref.fa"
params.outdir = "/scratch/user/gatk4/"
params.snpeff_db = "athalianaTair10"
params.pl = "illumina"
params.pm = "nextseq"
After activating your conda environment with Nextflow installed, run:
nextflow run main.nf
By default, Nextflow will look for a config file called nextflow.config inside the directory from which Nextflow was launched. You can specify a different config file on the command line using -c:
nextflow run main.nf -c your.config
Note: Parameters hard coded in the config can be overridden by arguments passed through the command line. See following scenario.
Scenario 2: Call the script providing all required parameters through the command line (after activating your Nextflow conda environment)
nextflow run main.nf \
--reads "/path/to/reads/*_n0{1,2}_*.fastq.gz" \
--ref "/path/to/ref.fa" \
--outdir "/scratch/user/gatk4/" \
--snpeff_db "athalianaTair10" \
--pl "illumina" \
--pm "illumina"
Note: You are able to set/override any and all parameters in this way.
Scenario 3: Call directly from GitHub
Nextflow allows you to run scripts hosted on GitHub directly. There is no need to download the script (main.nf) to your local computer. Rather, you can call the variant calling pipeline script directly from github by entering the following command:
nextflow run gencorefacility/variant-calling-pipeline-gatk4
In this scenario you still need the config file (which you can download from the git repo) to set your parameters (see Scenario 1), or pass parameters through the command line (see Scenario 2).
Scenario 4: Use with a Docker container
All software tools used in the pipeline have been packaged in a Docker container. You can instruct Nextflow to use a containerized Docker image for the analysis by passing the -with-docker or -with-singularity flags and the container. Calling the script directly from GitHub and telling it to use this docker image looks like this:
nextflow run gencorefacility/variant-calling-pipeline-gatk4 -with-docker gencorefacility/variant-calling-pipeline-gatk4
Launching Nextflow
Once Nextflow begins running, it will actively manage and monitor your jobs. If you’re working on the HPC at NYU, the corresponding nextflow.config file contains the line process.executor = 'slurm' which instructs Nextflow to submit jobs to SLURM. You can monitor your jobs in real time using watch and squeue:
watch squeue -u <netID>
Note: Since Nextflow actively manages your jobs, the session in which Nextflow is running must remain open. For example, if Nextflow is running and you lose power or network, Nextflow will be terminated. To avoid this, I recommend running Nextflow inside a tmux session, using nohup, or even submitting the nextflow script as a SLURM job. My preference is to use tmux (terminal multiplexer).
Enter the command tmux to enter a tmux session. Launch an interactive slurm session if you are on the NYU HPC. Activate your conda environment. Now, you can launch your Nextflow script.
“Detach” from the tmux session at any time by pressing “ctrl” + “B” together, then pressing “D” (for detach). If you ever lose power, connection, or detach, you can reattach your tmux session by typing tmux a. You will find your session running just as when you last left it.
If your Nextflow script does get interrupted for some reason, you can always resume the pipeline from the last successfully completed step using the -resume flag:
nextflow run main.nf -resume
Workflow Overview
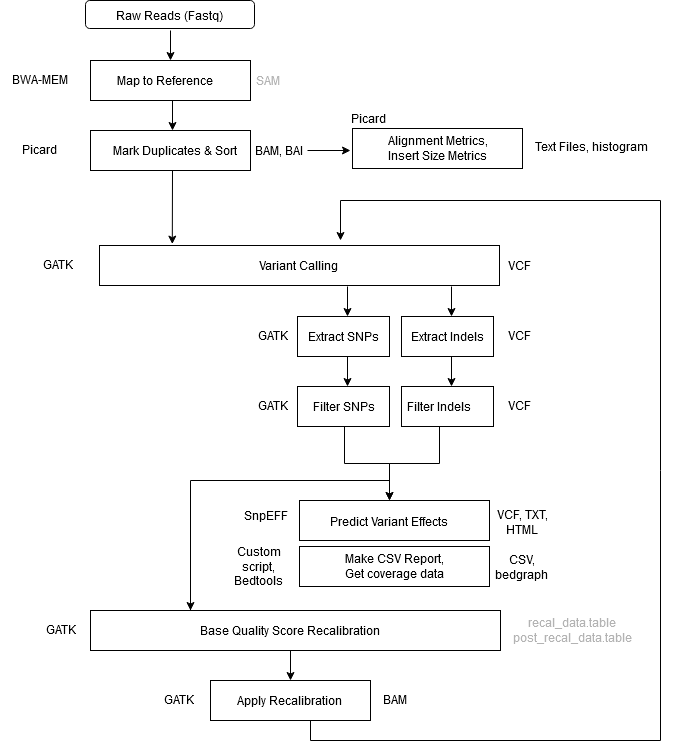
| Step 1 | Alignment – Map to Reference |
| Tool | BWA MEM |
| Input | .fastq files reference genome |
| Output | aligned_reads.sam |
| Notes | -Y tells BWA to use soft clipping for supplementary alignments-K tells BWA to process INT input bases in each batch regardless of nThreads (for reproducibility)Readgroup info is provided with the -R flag. This information is key for downstream GATK functionality. GATK will not work without a read group tag. |
| Command | bwa mem \ |
| Step 2 | Mark Duplicates + Sort |
| Tool | GATK4 MarkDuplicatesSpark |
| Input | aligned_reads.sam |
| Output | sorted_dedup_reads.bam sorted_dedup_reads.bam.bai dedup_metrics.txt |
| Notes | In GATK4, the Mark Duplicates and Sort Sam steps have been combined into one step using the MarkDuplicatesSpark tool. In addition, the BAM index file (.bai) is created as well by default. The tool is optimized to run on queryname-grouped alignments (that is, all reads with the same queryname are together in the input file). The output of BWA is query-grouped, however if provided coordinate-sorted alignments, the tool will spend additional time first queryname sorting the reads internally. Due to MarkDuplicatesSpark queryname-sorting coordinate-sorted inputs internally at the start, the tool produces identical results regardless of the input sort-order. That is, it will flag duplicates sets that include secondary, and supplementary and unmapped mate records no matter the sort-order of the input. This differs from how Picard MarkDuplicates behaves given the differently sorted inputs. (i.e. coordinate sorted vs queryname sorted). You don’t need a Spark cluster to run Spark-enabled GATK tools. If you’re working on a “normal” machine (even just a laptop) with multiple CPU cores, the GATK engine can still use Spark to create a virtual standalone cluster in place, and set it to take advantage of however many cores are available on the machine — or however many you choose to allocate. (https://gatk.broadinstitute.org/hc/en-us/articles/360035890591?id=11245) |
| Command | gatk \ |
| Step 3 | Collect Alignment & Insert Size Metrics |
| Tool | Picard Tools, R, Samtools |
| Input | sorted_dedup_reads.bam reference genome |
| Output | alignment_metrics.txt, insert_metrics.txt, insert_size_histogram.pdf, depth_out.txt |
| Command | java -jar picard.jar \java -jar picard.jar \CollectInsertSizeMetrics \samtools depth -a sorted_dedup_reads.bam > depth_out.txt |
| Step 4 | Call Variants |
| Tool | GATK4 |
| Input | sorted_dedup_reads.bam reference genome |
| Output | raw_variants.vcf |
| Notes | First round of variant calling. The variants identified in this step will be filtered and provided as input for Base Quality Score Recalibration (BQSR) |
| Command | gatk HaplotypeCaller \ |
| Step 5 | Extract SNPs & Indels |
| Tool | GATK4 |
| Input | raw_variants.vcf reference genome |
| Output | raw_indels.vcf raw_snps.vcf |
| Notes | This step separates SNPs and Indels so they can be processed and used independently |
| Command | gatk SelectVariants \ |
| Step 6 | Filter SNPs |
| Tool | GATK4 |
| Input | raw_snps.vcf reference genome |
| Output | filtered_snps.vcf filtered_snps.vcf.idx |
| Notes | The filters below are a good starting point provided by the Broad. You will need to experiment a little to find the values that are appropriate for your data, and to produce the tradeoff between sensitivity and specificity that is right for your analysis. QD < 2.0: This is the variant confidence (from the QUAL field) divided by the unfiltered depth of non-hom-ref samples. This annotation is intended to normalize the variant quality in order to avoid inflation caused when there is deep coverage. For filtering purposes it is better to use QD than either QUAL or DP directly. FS > 60.0: This is the Phred-scaled probability that there is strand bias at the site. Strand Bias tells us whether the alternate allele was seen more or less often on the forward or reverse strand than the reference allele. When there is little to no strand bias at the site, the FS value will be close to 0. MQ < 40.0: This is the root mean square mapping quality over all the reads at the site. Instead of the average mapping quality of the site, this annotation gives the square root of the average of the squares of the mapping qualities at the site. It is meant to include the standard deviation of the mapping qualities. Including the standard deviation allows us to include the variation in the dataset. A low standard deviation means the values are all close to the mean, whereas a high standard deviation means the values are all far from the mean.When the mapping qualities are good at a site, the MQ will be around 60. SOR > 4.0: This is another way to estimate strand bias using a test similar to the symmetric odds ratio test. SOR was created because FS tends to penalize variants that occur at the ends of exons. Reads at the ends of exons tend to only be covered by reads in one direction and FS gives those variants a bad score. SOR will take into account the ratios of reads that cover both alleles. MQRankSum < -8.0: Compares the mapping qualities of the reads supporting the reference allele and the alternate allele. ReadPosRankSum < -8.0: Compares whether the positions of the reference and alternate alleles are different within the reads. Seeing an allele only near the ends of reads is indicative of error, because that is where sequencers tend to make the most errors. Learn more about hard filtering: https://gatk.broadinstitute.org/hc/en-us/articles/360035890471-Hard-filtering-germline-short-variants Note: SNPs which are ‘filtered out’ at this step will remain in the filtered_snps.vcf file, however they will be marked as ‘_filter’, while SNPs which passed the filter will be marked as ‘PASS’. We need to extract and provide only the passing SNPs to the BQSR tool, we do this in the next step (step 9). |
| Command | gatk VariantFiltration \ |
| Step 7 | Filter Indels |
| Tool | GATK4 |
| Input | raw_indels.vcf reference genome |
| Output | filtered_indels.vcf filtered_indels.vcf.idx |
| Notes | The filters below are a good starting point provided by the Broad. You will need to experiment a little to find the values that are appropriate for your data, and to produce the tradeoff between sensitivity and specificity that is right for your analysis. QD < 2.0: This is the variant confidence (from the QUAL field) divided by the unfiltered depth of non-hom-ref samples. This annotation is intended to normalize the variant quality in order to avoid inflation caused when there is deep coverage. For filtering purposes it is better to use QD than either QUAL or DP directly. FS > 200.0: This is the Phred-scaled probability that there is strand bias at the site. Strand Bias tells us whether the alternate allele was seen more or less often on the forward or reverse strand than the reference allele. When there is little to no strand bias at the site, the FS value will be close to 0. SOR > 10.0: This is another way to estimate strand bias using a test similar to the symmetric odds ratio test. SOR was created because FS tends to penalize variants that occur at the ends of exons. Reads at the ends of exons tend to only be covered by reads in one direction and FS gives those variants a bad score. SOR will take into account the ratios of reads that cover both alleles. Learn more about hard filtering: https://gatk.broadinstitute.org/hc/en-us/articles/360035890471-Hard-filtering-germline-short-variants Note: Indels which are ‘filtered out’ at this step will remain in the filtered_snps.vcf file, however they will be marked as ‘_filter’, while SNPs which passed the filter will be marked as ‘PASS’. We need to extract and provide only the passing indels to the BQSR tool, we do this next. |
| Command | gatk VariantFiltration \ |
| Step 8 | Exclude Filtered Variants |
| Tool | GATK4 |
| Input | filtered_snps.vcf filtered_indels.vcf |
| Output | bqsr_snps.vcf bqsr_indels.vcf |
| Notes | We need to extract only the passing variants and provide this as input to BQSR (next step). |
| Command | gatk SelectVariants \ --exclude-filtered \ -O bqsr_indels.vcf |
| Step 9 | Base Quality Score Recalibration (BQSR) #1 |
| Tool | GATK4 |
| Input | sorted_dedup_reads.bam (from step 2) bqsr_snps.vcf bqsr_indels.vcf reference genome |
| Output | recal_data.table |
| Notes | BQSR is performed twice. The second pass is optional, only required to produce a recalibration report. |
| Command | gatk BaseRecalibrator \ -R ref.fa \ |
| Step 10 | Apply BQSR |
| Tool | GATK4 |
| Input | recal_data.table sorted_dedup_reads.bam reference genome |
| Output | recal_reads.bam |
| Notes | This step applies the recalibration computed in the first BQSR step to the bam file. This recalibrated bam file is now analysis-ready. |
| Command | gatk ApplyBQSR \ -R ref.fa \ |
| Step 11 | Base Quality Score Recalibration (BQSR) #2 |
| Tool | GATK4 |
| Input | recal_reads.bam bqsr_snps.vcf bqsr_indels.vcf reference genome |
| Output | post_recal_data.table |
| Notes | This round of BQSR is optional. It is required if you want to produce a recalibration report with the Analyze Covariates step (next). For this round of BQSR, you provide the recalibrated reads obtained from the Apply BQSR step above as input. |
| Command | gatk BaseRecalibrator \ -R ref.fa \ |
| Step 12 | Analyze Covariates |
| Tool | GATK4 |
| Input | recal_data.table post_recal_data.table |
| Output | recalibration_plots.pdf |
| Notes | This step produces a recalibration report based on the output from the two BQSR runs |
| Command | gatk AnalyzeCovariates \ |
| Step 13 | Call Variants |
| Tool | GATK4 |
| Input | recal_reads.bam reference genome |
| Output | raw_variants_recal.vcf |
| Notes | Second round of variant calling performed using recalibrated (analysis-ready) bam |
| Command | gatk HaplotypeCaller \ |
| Step 14 | Extract SNPs & Indels |
| Tool | GATK4 |
| Input | raw_variants_recal.vcf reference genome |
| Output | raw_indels_recal.vcfraw_snps_recal.vcf |
| Notes | This step separates SNPs and Indels so they can be processed and analyzed independently |
| Command | gatk SelectVariants \ -R ref.fa \ -R ref.fa \ |
| Step 15 | Filter SNPs |
| Tool | GATK4 |
| Input | raw_snps_recal.vcf reference genome |
| Output | filtered_snps_final.vcf filtered_snps_final.vcf.idx |
| Notes | The filters below are a good starting point provided by the Broad. You will need to experiment a little to find the values that are appropriate for your data, and to produce the tradeoff between sensitivity and specificity that is right for your analysis. QD < 2.0: This is the variant confidence (from the QUAL field) divided by the unfiltered depth of non-hom-ref samples. This annotation is intended to normalize the variant quality in order to avoid inflation caused when there is deep coverage. For filtering purposes it is better to use QD than either QUAL or DP directly. FS > 60.0: This is the Phred-scaled probability that there is strand bias at the site. Strand Bias tells us whether the alternate allele was seen more or less often on the forward or reverse strand than the reference allele. When there is little to no strand bias at the site, the FS value will be close to 0. MQ < 40.0: This is the root mean square mapping quality over all the reads at the site. Instead of the average mapping quality of the site, this annotation gives the square root of the average of the squares of the mapping qualities at the site. It is meant to include the standard deviation of the mapping qualities. Including the standard deviation allows us to include the variation in the dataset. A low standard deviation means the values are all close to the mean, whereas a high standard deviation means the values are all far from the mean.When the mapping qualities are good at a site, the MQ will be around 60. SOR > 4.0: This is another way to estimate strand bias using a test similar to the symmetric odds ratio test. SOR was created because FS tends to penalize variants that occur at the ends of exons. Reads at the ends of exons tend to only be covered by reads in one direction and FS gives those variants a bad score. SOR will take into account the ratios of reads that cover both alleles. MQRankSum < -8.0: Compares the mapping qualities of the reads supporting the reference allele and the alternate allele. ReadPosRankSum < -8.0: Compares whether the positions of the reference and alternate alleles are different within the reads. Seeing an allele only near the ends of reads is indicative of error, because that is where sequencers tend to make the most errors. Learn more about hard filtering: https://gatk.broadinstitute.org/hc/en-us/articles/360035890471-Hard-filtering-germline-short-variants Note: SNPs which are ‘filtered out’ at this step will remain in the filtered_snps.vcf file, however they will be marked as ‘_filter’, while SNPs which passed the filter will be marked as ‘PASS’. |
| Command | gatk VariantFiltration \ |
| Step 16 | Filter Indels |
| Tool | GATK4 |
| Input | raw_indels_recal.vcf reference genome |
| Output | filtered_indels_final.vcf filtered_indels_final.vcf.idx |
| Notes | The filters below are a good starting point provided by the Broad. You will need to experiment a little to find the values that are appropriate for your data, and to produce the tradeoff between sensitivity and specificity that is right for your analysis. QD < 2.0: This is the variant confidence (from the QUAL field) divided by the unfiltered depth of non-hom-ref samples. This annotation is intended to normalize the variant quality in order to avoid inflation caused when there is deep coverage. For filtering purposes it is better to use QD than either QUAL or DP directly. FS > 200.0: This is the Phred-scaled probability that there is strand bias at the site. Strand Bias tells us whether the alternate allele was seen more or less often on the forward or reverse strand than the reference allele. When there is little to no strand bias at the site, the FS value will be close to 0. SOR > 10.0: This is another way to estimate strand bias using a test similar to the symmetric odds ratio test. SOR was created because FS tends to penalize variants that occur at the ends of exons. Reads at the ends of exons tend to only be covered by reads in one direction and FS gives those variants a bad score. SOR will take into account the ratios of reads that cover both alleles. Learn more about hard filtering: https://gatk.broadinstitute.org/hc/en-us/articles/360035890471-Hard-filtering-germline-short-variants Note: Indels which are ‘filtered out’ at this step will remain in the filtered_snps.vcf file, however they will be marked as ‘_filter’, while SNPs which passed the filter will be marked as ‘PASS’. |
| Command | gatk VariantFiltration \ |
| Step 17 | Annotate SNPs and Predict Effects |
| Tool | SnpEff |
| Input | filtered_snps_final.vcf |
| Output | filtered_snps_final.ann.vcf snpeff_summary.html snpEff_genes.txt |
| Command | java -jar snpEff.jar -v \ |
| Step 18 | Compile Statistics |
| Tool | parse_metrics.sh (in /bin) |
| Input | sample_id_alignment_metrics.txt sample_id_insert_metrics.txt sample_id_dedup_metrics.txt sample_id_raw_snps.vcf sample_id_filtered_snps.vcf sample_id_raw_snps_recal.vcf sample_id_filtered_snps_final.vcf sample_id_depth_out.txt |
| Output | report.csv |
| Command | parse_metrics.sh sample_id > sample_id_report.csv |
| Notes | A single report file is generated with summary statistics for each library processed containing the following pieces of information: # of Reads # of Aligned Reads % Aligned # Aligned Bases Read Length % Paired % Duplicate Mean Insert Size # SNPs, # Filtered SNPs # SNPs after BQSR, # Filtered SNPs after BQSR Average Coverage Give the script a sample id and it will look for the corresponding sample_id_* files (listed in the input section above) and print the statistics and header to stdout. |


62 Comments
Eric Borenstein · 2020-04-03 at 1:34 pm
This workflow can also be ran as an SBATCH rather than interactively. The SBATCH options to change would be job-name, output, and possibly time. The resources set in SBATCH are only for the job controller nextflow and not the actual compute, so no need to increase. The resources for your compute would be set in the config file given. Job process can be monitored in the slurm output file you set.
#!/bin/bash
#SBATCH –nodes=1
#SBATCH –ntasks-per-node=1
#SBATCH –cpus-per-task=2
#SBATCH –time=5:00:00
#SBATCH –mem=2GB
#SBATCH –job-name=myJob
#SBATCH –output=slurm_%j.out
module purge
module load nextflow/20.10.0
nextflow run main.nf -c your.config
MARION · 2020-05-01 at 10:18 am
Thank you so much for this elaborate workflow Mohammed. However I am continuously getting this error nextflow.exception.AbortOperationException: Not a valid project name: main.nf when I run nextflow run main.nf. Thank you in advance!
Mohammed Khalfan · 2020-05-04 at 6:03 pm
This error means there is no main.nf file in your working directory. You should change into the directory where the main.nf script is located and then run nextflow run main.nf.
Darach · 2020-05-03 at 1:52 pm
Glad to see you could use that filter/tap trick, and the adoption of nextflow generally. Cleanly written.
Does NYU HPC let you use Docker?
Mohammed Khalfan · 2020-05-04 at 6:05 pm
Thank you Darach, I’m really liking nextflow. There are a couple other workflows on our git too. Have you had the opportunity to test out the modules feature? I’m interested to try that next.
We use Singularity on the HPC which supports the use of Docker containers.
Darach · 2020-05-04 at 7:30 pm
No, I haven’t seen modules yet. It looks like a really smart idea. I look forward to what y’all write in that; I know that’ll be really cleanly done.
Ah, so it makes sense to write it as a Docker recipe, because then that’s more flexible for users in both platforms. Presumably folks using cloud-computing will appreciate that.
I’m going to copy the way you’re writing channel operators in the actual input field. I hadn’t thought to do that, and I think that style is a much cleaner way of organizing nextflow. Thanks for sharing your code with everyone, for direct use and inspiration.
Mohammed Khalfan · 2020-05-06 at 10:22 pm
Thank you for the kind words Darach, I value your feedback.
Marion · 2020-05-07 at 1:27 pm
Hi Mohammed, I got an error at the markDuplicatesSpark step caused by: java.lang.IllegalStateException: Duplicate key A00431:16:H5JTVDSXX:1:1101:27308:32972 (attempted merging values -1 and -1). I wonder what it is all about, I have tried to figure out how to go about this and failed. Thank you in advance.
Marion · 2020-05-08 at 9:49 am
Was able to resolve this by upgrading my GATK version from V4.0.4.0 to V4.0.5.0. Thank you!
Crystal · 2020-05-21 at 5:01 pm
Hi there,
Iam continuosly getting this error below;
Process `haplotypeCaller (1)` terminated with an error exit status (2)
A USER ERROR has occurred: Traversal by intervals was requested but some input files are not indexed.
Please index all input files:
samtools index ………………._sorted_dedup.bam
Set the system property GATK_STACKTRACE_ON_USER_EXCEPTION (–java-options ‘-DGATK_STACKTRACE_ON_USER_EXCEPTION=true’) to print the stack trace.
I attempted to index it manually with samtools however when I resumed the process. it couldn’t locate my indexed bam files.
Your help is much appreciated.
Yoshi · 2020-07-08 at 12:52 am
Hi, Mohammed. Thank you for the great pipeline.
I would like to use this pipeline for strain tracking of pathogenic bacteria in our society based on SNVs, and I have one question.
How can we mask (exclude) some genomic regions from the reference genome ? To go around false-positive SNVs, we usually exclude repetitive region as short reads are not correctly mapped to these regions. We also exclude dense SNVs that are due to incorrect mapping using filtering of window size 12 bp. Can we do this as well ? Many thanks for your kind support.
Mohammed Khalfan · 2020-09-10 at 6:39 pm
Hi Yoshi,
You can use provide haplotypecaller with an intervals file with the -L option, and it will work on only the regions defined in this file. See https://gatk.broadinstitute.org/hc/en-us/articles/360035531852-Intervals-and-interval-lists.
Diego · 2020-08-25 at 9:35 pm
Hi Mohammed, thanks so much for this comprehensive pipeline. I was wondering, do I need to cat and trim (with trimmomatic) my raw reads prior to starting the pipeline?
Mohammed Khalfan · 2020-08-27 at 3:17 pm
Hi Diego, there is no strict rule, but it’s probably better to do this.
Niyomi · 2020-09-01 at 8:38 pm
Is this pipeline for an individual sample? I don’t see any merging steps anywhere. At what point should I merge the samples if I were to use this? BAM files or the VCF’s?
Mohammed Khalfan · 2020-09-02 at 6:45 pm
Hi Niyomi,
This pipeline operates HaplotypeCaller in its default mode on a single sample. If you would like to do joint genotyping for multiple samples, the pipeline is a little different. You would need to add the
-ERC GVCFoption to HaplotypeCaller to generate an intermediate GVCF, and then run gatk GenotypeGVCFs using the intermediary GVCFs as input.Niyomi · 2020-09-03 at 9:45 am
Thank you for your reply. Yes, I am familiar with the -ERC GVCF. My question is, do you use this pipeline to process each sample individually or do you merge samples at any point? In other words, do I follow this pipeline 250 times for 250 samples? Thank you in advance.
Mohammed Khalfan · 2020-09-10 at 2:35 pm
This pipeline operates on each sample individually, it does not merge samples at any point, so yes you would follow the pipeline 250 times for 250 samples. The nextflow script takes care of this.
Ashok · 2021-04-19 at 12:46 am
Hi Niyomi,
I am running it on multiple human genome sequence samples (bam files). Could you please share your experience of working to adopt this script to multiple samples?
Julia Xing · 2020-09-10 at 1:16 am
Which steps should I customize if I use single read sequencing instead of paired end run? Thanks a lot!
Mohammed Khalfan · 2020-09-10 at 2:38 pm
Primarily the alignment step, and the way input reads are parsed by nextflow. CollectInsertSizeMetrics in the parse metrics step will fail, as will this part of the final QC step.
XiaTian · 2020-11-09 at 9:33 pm
Thanks for your sharing. Still I have a basic question. When we use GATK HaplotypeCaller, it costs a lot of time (800M genome, 15* coverage, 5-7days, per sample). Should I set “NUMBERTHREADS” or “Xmx96g”in this step?
Nathalia · 2020-11-16 at 9:40 am
Hello, thanks for this pipeline, it’s very helpful!
I was able to substitute bwa for minimap2 command, however with minimap2 I need to use picard AddOrReplaceReadGroups command before gatk MarkDuplicatesSpark. Can I add another process in main.nf? I’ve tried, but no success so far.
Mohammed Khalfan · 2020-12-02 at 5:54 pm
Hi Nathalia, you can add another process but you will also need to setup the input channel to send data into that process, and setup the channel to send output from the new process to MarkDuplicatesSpark.
zeeshan Fazal · 2020-12-02 at 1:32 pm
Hi Mohammed
I am trying to do variant calling and I have human cell lines. I am really confused that why your pipeline doesn’t not use Goldstandard SNPs and dbSNPs like in GATK? And Can i Run this for human data?
Mohammed Khalfan · 2020-12-02 at 5:58 pm
Hi Zeehan,
This workflow is adapted for organisms that do not have gold standard SNPs available. If you’re working with human data I would check out broads official workflows such as this one: https://github.com/gatk-workflows/gatk4-germline-snps-indels
zeeshan Fazal · 2020-12-04 at 3:02 pm
Thanks Mohammed!
Ashok · 2021-04-19 at 12:49 am
Hi Zeeshan,
I am running it on multiple human genome sequence samples (bam files) where we have multiple gold standard resources. Am I supposed to mention them as parameters and use them just like reference file? Could you please explain what you followed?
Many thanks!
xiaochen · 2021-03-21 at 4:34 am
Hi,
This pipeline is for diploid or haploid organism? I used this pipeline in haploid and found many AF=0.5 snps or indels ? Can you help me ?Thank you .
Harish · 2021-07-13 at 12:22 pm
The reason for that is GATK by default tends to assume that the variants are being called for a diploid organism. You can change that by using the ‘–ploidy ‘ switch in HaplotypeCaller.
VIVEK RUHELA · 2020-12-14 at 1:56 am
Hello,
My question is that why the BQSR step is after haplotype variant calling. I think all the BAM record post-processing (like sort + remote duplicates + BQSR) should be before variant calling. Let me know if this is wrong? Can I follow the same step for somatic mutation identification (using mutect2 in place of gatk haplotype caller)?
Matt LaBella · 2021-02-25 at 12:10 pm
Hi Mohammed,
First thank you for providing this pipeline publicly, I’m learning a lot! I ran into an error at the getMetrics step and I wasn’t sure if I just made a simple mistake. The error is:
.command.sh: line 2: PICARD_JAR: unbound variable
Thanks again, Matt
Jaikrishna · 2021-11-26 at 1:51 pm
Have been facing the same issue. Would be great if someone can share a fix
Mohammed Khalfan · 2021-12-08 at 6:26 pm
Are you using the container? the $PICARD_JAR variable exists within the container.
Vinoy · 2022-03-09 at 3:51 pm
I am getting the same error. Any advice to fix it. Thanks
Hassan Badrane · 2021-06-14 at 11:37 am
I would also like to thank Mohammed for this very useful pipeline. I’m a beginner and I knew it will take me a while to build my own pipeline, so having this very elaborated pipeline was wonderful and it saved me a lot of time. I successfully run the pipeline on close to 100 yeast strains, with a few customizations. I run the pipeline directly using the preinstalled Nextflow module, and not by creating the nextflow env, which was a bit difficult as it generated many errors.
In case this can help someone, one thing that helped me in the process is the configuration of Nextflow to run as a slurm. This can be done by editing the “nextflow.config” file to something like this:
// Nextflow Parameters
process {
executor = ‘slurm’
}
Harish · 2021-07-09 at 6:23 am
Hi! Thanks for the pipeline. It was a good step for me to start learning Nextflow!
What I was wondering is, how do you cache the results from MarkDuplicates? Everytime the pipeline fails, I need to start from Marking the duplicates.
Mohammed Khalfan · 2021-12-08 at 6:25 pm
You would need to *delete* the lines (not comment out) where you make the temp directory and remove it (first and last lines in the process), and remove the parameter that tells GATK to use the tmpdir. The use of a temporary directory, and the way it is named when created, results in the underlying code being different every time the script it run and this causes the pipeline to resume from the step. The temporary directory was introduced originally because the compute nodes where the processes run were running out of space when working with larger datasets.
Harish · 2021-07-13 at 12:27 pm
Hi Mohammed!
I was wondering why doesn’t subsequent steps after alignment cache the results? I keep having to mark the dups each time from alignments. Is there any way to force trigger it? cache deep/true/lenient is not picking up the files as does resume.
Ram · 2021-07-13 at 6:33 pm
Hi Mohammed Khalfan, I would like to request you to add scripts for the Mutect2 (to call somatic variants) and at which step (parallel to Haplotype Caller) it should be incorporated in the pipeline and what steps should be done next.
Thanks in advance !!!
Francesco · 2021-08-26 at 12:21 pm
Dear Mohammed, I am experimenting with your workflow from outside NYU. As a test run, I am working with a short reference sequence, and only 5% of my reads are actually mapping there. In order to avoid huge sam/bam files, would you advise to filter the initial sam for mapped reads only (inverse-grepping AS:i:0 or, in order to conserve pairs, based on the ‘2’ bitwise flag)?
By the way, thanks for your elaborate explanations, I will try to make good use of them.
Mohammed Khalfan · 2021-12-08 at 6:31 pm
You could do that, but the pipeline takes fastq as input, so you would then need to convert the bamback to fastq, then use that as input. If you do decide to do that, you can use `samtools view -F 4 file.bam` to get mapped reads only from a bam file.
Tiago · 2021-11-02 at 2:00 pm
Hello, quando rodo:
bash /guest/triatominae/tiagob/snp/m1/pasta/pasta/gatk-4.2.2.0/parse_metric.sh sample_id sample_id_ > sample_id_report.csv
não gera pra mim . sample_id_report.csv, o que pode estar errado?
input:
sample_id_alignment_metrics.txt
sample_id_depth_out.txt
sample_id_filtered_snps.vcf
sample_id_filtered_snps_final.vcf
sample_id_insert_metrics.txt
sample_id_raw_snps.vcf
sample_id_raw_snps_recal.vcf
parse_metric.sh >>>
#!/bin/bash
ID=$1
input=${ID}_alignment_metrics.txt
while read line
do
if [[ $line == PAIR* ]];then
ALIGNMENT_METRICS=$(echo $line | cut -d’ ‘ -f2,6,7,10,16,18 | tr ‘ ‘ ‘,’)
fi
done < $input
re='^[0-9]+([.][0-9]+)?$'
input=${ID}_insert_metrics.txt
while read line
do
MEAN_INSERT_SIZE=$(echo $line | cut -d' ' -f5)
if [[ $MEAN_INSERT_SIZE =~ $re ]];then
break
fi
done > ../report.csv
Tiago · 2021-11-02 at 2:03 pm
Tiago · 2021-11-02 at 2:00 pm
Hello, when I run:
bash /guest/triatominae/tiagob/snp/m1/pasta/pasta/gatk-4.2.2.0/parse_metric.sh sample_id sample_id_ > sample_id_report.csv
#—–#—–#—–
don’t generate for me = sample_id_report.csv, what can be wrong?
#—–#—–#—–
input file:
sample_id_alignment_metrics.txt
sample_id_depth_out.txt
sample_id_filtered_snps.vcf
sample_id_filtered_snps_final.vcf
sample_id_insert_metrics.txt
sample_id_raw_snps.vcf
sample_id_raw_snps_recal.vcf
#—–
parse_metric.sh (code)
#!/bin/bash
ID=$1
input=${ID}_alignment_metrics.txt
while read line
do
if [[ $line == PAIR* ]];then
ALIGNMENT_METRICS=$(echo $line | cut -d’ ‘ -f2,6,7,10,16,18 | tr ‘ ‘ ‘,’)
fi
done ../report.csv
SINUMOL GEORGE · 2021-12-08 at 10:10 am
I want to run GATk to filter somatic variants? Please explain the workflow for human somatic variant calling.
Thanks,
Sinumol
Mohammed Khalfan · 2021-12-08 at 6:33 pm
This pipeline uses HaplotypeCaller which is meant for germline variants. Try mutect2 for somatic variants.
sinu · 2023-04-27 at 2:18 am
Could you please provide that workflow as well?
Joyce Wangari · 2022-01-13 at 10:53 am
Hello, Mohammed I am trying to run the pipeline and it is giving me an error:
Command executed:
bwa mem -K 100000000 -v 3 -t 1 -Y -R “@RG\tID:Tryp-B87_S2\tLB:Tryp-B87_S2\tPL:illumina\tPM:illumina\tSM:Tryp-B87_S2” -M {/node/cohort4/joyce/variant_calling/variant-calling-pipeline-gatk4/snpEff/data/genomes/IL3000.fa} Tryp-B87_S2_1.fastq Tryp-B87_S2_2.fastq > Tryp-B87_S2_aligned_reads.sam
Command exit status:
1
Command output:
(empty)
Command error:
[E::bwa_idx_load_from_disk] fail to locate the index files
What should i do please?
Mohammed Khalfan · 2022-02-11 at 6:10 pm
Need to make sure the BWA index files are in the same directory as the reference FASTA file.
Jellyfish · 2022-01-16 at 9:13 am
hey dear Mohammed Khalfan~~~///(^v^)\\\~~~,
thanks a lot for this amazing tutorial,
when I run the step 4,there would always be like
“java.lang.IllegalArgumentException: Something went wrong with sequence dictionary detection, check that reference has a valid sequence dictionary ”
I went though the first 3 steps smoothly ,and I’m pretty sure I have set everything all right,how can I handle this?
Mohammed Khalfan · 2022-02-11 at 6:09 pm
Need to make sure that the sequence dictionary (.dict file, built using Picard CreateSequenceDictionary tool) is in the same directory as the reference FASTA file.
Harun · 2022-03-22 at 1:37 pm
Hi Mohammed,
thanks for this pipeline, it’s very helpful for understanding of the each step!
All the best
Ziv · 2022-11-20 at 4:03 pm
Hi.
Is it possible to modify this protocol to single-end sequences?
Cassandra Buzby · 2023-09-05 at 2:24 pm
Hi Ziv, I have adjusted the pipeline for my own single-end sequences if you’d like me to send it over.
Joel · 2023-01-28 at 5:30 pm
Hi Mohammed Khalfan,
Thanks for this usefull tutorial, just wondering why you only do SNPeff on the SNPS and not on the indels? Or in other words merge SNP vcf and indels and than call SNPeff. Although indels are not in the name ‘SNPeff’ as far as I know it could handle indel variants and annotate them accordingly.
Thanks again, Joel
Kenneth · 2023-10-09 at 3:24 pm
Hi Mohammed,
Quick question about a workflow error I receive. The workflow gets stuck on step 2 MarkDuplicatesSpark with error: markDuplicatesSpark (1)’ terminated with an error exit status (2). A USER ERROR has occurred: Failed to load reads from _aligned_reads.sam Caused by:Input path does not exist: file:_aligned_reads.sam
I’m confused by this error since the script creates the _aligned_reads.sam file in the nextflow working directory. Why can’t the file be found?
Any help is greatly appreciated,
Kenneth
Stefany · 2024-06-26 at 6:02 pm
Hello!
I`m running this pipeline in Coffea mutants, however I’m not sure if instead should have used Mutect2. Also, I’m not familiar with this sort of analysis, therefore I’m not really sure what to expect for the final output (*.ann.vcf file). Could you post a tinny example of how it should look like if ran successfully?
In addition, could you share de parse_metrics.sh script?
Best wishes,
Stefany
Sakshi · 2024-07-29 at 9:20 am
Hi, I want to perform variant calling in Soybean. First question is -will the script be helpful for plant genomes?
Second, – how can we detect variants for two samples -resistant vs susceptible. and can we merge the two samples and then combinedly call variants?
perla · 2024-08-15 at 12:19 pm
Hi Mohammed,
Im trying to run the script on step 5:
gatk SelectVariants \
-R ref.fa \
-V raw_variants.vcf \
-selectType SNP \
-o raw_snps.vcf
but when i use it it gives me this error: A USER ERROR has occurred: -selectType is not a recognized option
sorry if my question is simple or obvious. im very new into this topic thank you.
Mohammed Khalfan · 2024-08-15 at 3:12 pm
It should be
-select-type SNP, I’ve updated the post.Sineshaw Legese · 2024-09-26 at 7:36 am
Hello Mohammed, I have multiple VCF files from various samples. How can I prepare these samples for the Bootstrapping step?
Akriti · 2024-10-08 at 9:01 am
Hi,
Great tutorial. I am using this pipeline to do variant calling on a diploid S.cervisiae. When I used it default params.hc_config = “-ploidy 1, it worked, but when I changed it to 2. I am getting this error:
Error executing process > ‘qualimap (1)’
Caused by:
Process `qualimap (1)` terminated with an error exit status (140)
Any help would be great
Best,
Akriti