Update: This pipeline is now deprecated. See the updated version of the variant calling pipeline using GATK4.
Identifying genomic variants, such as single nucleotide polymorphisms (SNPs) and DNA insertions and deletions (indels), can play an important role in scientific discovery. To this end, a pipeline has been developed to allow researchers at the CGSB to rapidly identify and annotate variants. The pipeline employs the Genome Analysis Toolkit (GATK) to perform variant calling and is based on the best practices for variant discovery analysis outlined by the Broad Institute. Once SNPs have been identified, SnpEff is utilized to annotate and predict the effects of the variants.
Full List of Tools Used in this Pipeline:
- GATK
- BWA
- Picard
- Samtools
- Bedtools
- SnpEff
- R (dependency for some GATK and Picard steps)
Script Location:
prince:/scratch/work/cgsb/scripts/variant_calling/pipeline.sh
https://github.com/gencorefacility/variant-calling-pipeline
How to use this script:
- Create a new directory for this project in your home folder on scratch
cd /scratch/<netID>/mkdir variant-callingcd variant-calling - Copy pipeline.sh into your project directory
cp /scratch/work/cgsb/scripts/variant-calling/pipeline.sh . - The pipeline requires six input values. The values can be hard coded into the script, or provided as command line arguments. They are found at the very top of the script.
- DIR: The directory with your FastQ libraries to be processed. This should be located in /scratch/cgsb/gencore/out/<PI>/
- REF: Reference genome to use. This should be located in the Shared Genome Resource (/scratch/work/cgsb/reference_genomes/). Selecting a reference genome from within the Shared Genome Resource ensures that all index files and reference dictionaries required by BWA, Picard, GATK, etc. are available.
- SNPEFF_DB: The name of the SnpEff database to use. To determine the name of the SnpEff DB to use, issue the following command:
java -jar /share/apps/snpeff/4.1g/snpEff.jar databases | grep -i SPECIES_NAMEFrom the list of returned results, select the database you wish to use. The value of the first column is the value to input for SNPEFF_DB.Example:
java -jar /share/apps/snpeff/4.1g/snpEff.jar databases | grep -i thalianaOutput:
athalianaTair10 Arabidopsis_Thaliana http://downloads.sourceforge.net/...Then:
SNPEFF_DB='athalianaTair10'Note: If your organism is not available in SnpEff, it is possible to create a custom SnpEff Database if a reference fasta and gff file are available.
- PL: Platform. Ex: illumina
- PM: Machine. Ex: nextseq
- EMAIL: Your email address. Used to report PBS job failures
In the example below, I have hard coded the parameters in the script:
DIR='/data/cgsb/gencore/out/Gresham/2015-10-23_HK5NHBGXX/lib1-26/'REF='/scratch/work/cgsb/reference_genomes/Public/Fungi/Saccharomyces_cerevisiae/GCF_000146045.2_R64/GCF_000146045.2_R64_genomic.fna'SNPEFF_DB='GCF_000146045.2_R64'PL='illumina'PM='nextseq'EMAIL='some1@nyu.edu' # Not a real email address
- Run the script. If you hard coded the parameters in the script as shown in the example above, no additional parameters need to be provided. Simply run:
sh pipeline.sh - Optional: Monitor the jobs
watch -d qstat -u <netID>
Overview of the pipeline
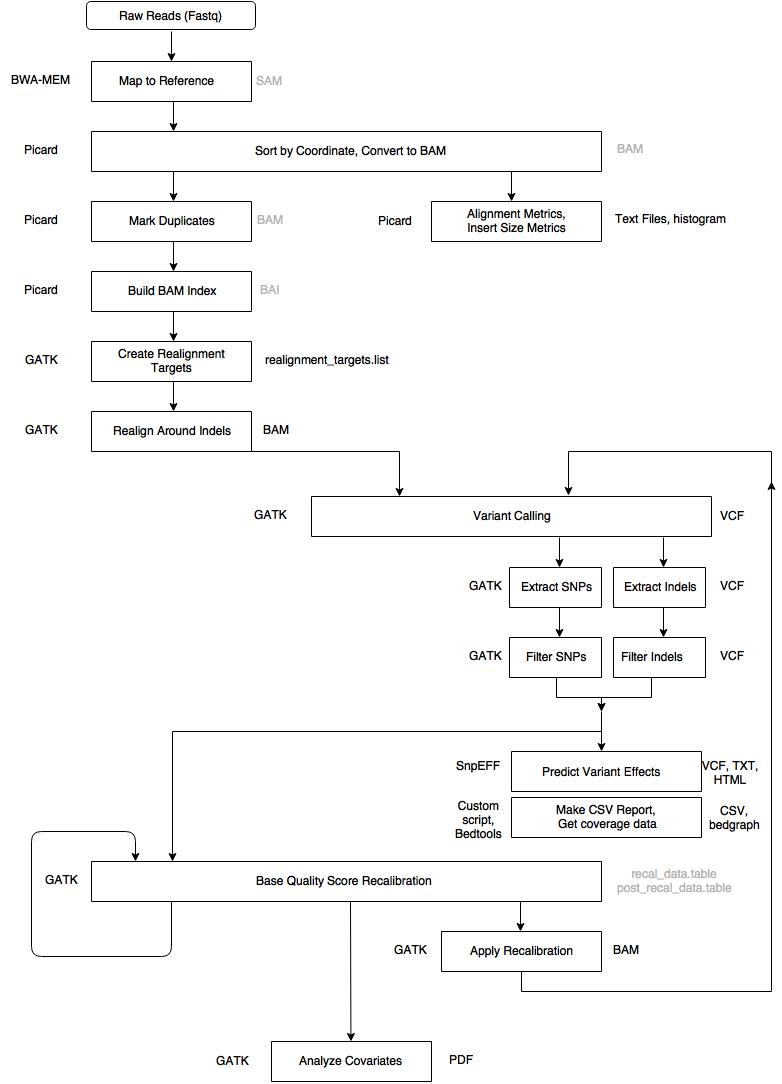
Important Notes:
- The pipeline assumes no known variants are available for the Base Quality Score Recalibration step and as such bootsraps a set of SNPs and Indels to provide as input at this step. Contact me if a list of high quality known variants is available for your organism as this data can improve the quality of the output
- The current script is designed to work with Paired End data
- Due to HPC queue limits on mercer, the pipeline can run on a maximum of 25 libraries at a time
- If you need to run the pipeline on more than 25 libraries or on Single End reads, or have any other questions, please contact me
Walk through
| Step 1 | Alignment – Map to Reference |
| Tool | BWA MEM |
| Input | .fastq files, reference genome |
| Output |
aligned_reads.sam* *Intermediary file, removed from final output |
| Notes |
Need to provide the -M flag to BWA, this tells it to consider split reads as secondary, need this for GATK variant calling/Picard support. Alternate alignment tools: Bowtie2, Novoalign Readgroup info is provided with the -R flag. This information is key for downstream GATK functionality. GATK will not work without a read group tag. |
| Command | bwa mem -M -R '@RG\tID:sample_1\tLB:sample_1\tPL:ILLUMINA\tPM:HISEQ\tSM:sample_1' ref input_1 input_2 > aligned_reads.sam |
| Step 2 | Sort SAM file by coordinate, convert to BAM |
| Tool | Picard Tools |
| Input | aligned_reads.sam |
| Output |
sorted_reads.bam* *Intermediary file, removed from final output |
| Command | java -jar picard.jar SortSam INPUT=aligned_reads.sam OUTPUT=sorted_reads.bam SORT_ORDER=coordinate |
| Step 3 | Collect Alignment & Insert Size Metrics |
| Tool | Picard Tools, R, Samtools |
| Input | sorted_reads.bam, reference genome |
| Output | alignment_metrics.txt, insert_metrics.txt, insert_size_histogram.pdf, depth_out.txt |
| Command |
|
| Step 4 | Mark Duplicates |
| Tool | Picard Tools |
| Input | sorted_reads.bam |
| Output |
dedup_reads.bam* metrics.txt *Intermediary file, removed from final output |
| Command | java -jar picard.jar MarkDuplicates INPUT=sorted_reads.bam OUTPUT=dedup_reads.bam METRICS_FILE=metrics.txt |
| Step 5 | Build BAM Index |
| Tool | Picard Tools |
| Input | dedup_reads.bam |
| Output |
dedup_reads.bai* *Intermediary file, removed from final output |
| Command | java -jar picard.jar BuildBamIndex INPUT=dedup_reads.bam |
| Step 6 | Create Realignment Targets |
| Tool | GATK |
| Input | dedup_reads.bam, reference genome |
| Output | realignment_targets.list |
| Notes | This is the first step in a two-step process of realigning around indels |
| Commad | java -jar GenomeAnalysisTK.jar -T RealignerTargetCreator -R ref -I dedup_reads.bam -o realignment_targets.list |
| Step 7 | Realign Indels |
| Tool | GATK |
| Input | dedup_reads.bam, realignment_targets.list, reference genome |
| Output | realigned_reads.bam |
| Notes | This step performs the realignment around the indels which were identified in the previous step (the ‘realignment targets’) |
| Command | java -jar GenomeAnalysisTK.jar -T IndelRealigner -R ref -I dedup_reads.bam -targetIntervals realignment_targets.list -o realigned_reads.bam |
| Step 8 | Call Variants |
| Tool | GATK |
| Input | realigned_reads.bam, reference genome |
| Output | raw_variants.vcf |
| Notes | First round of variant calling. The variants identified in this step will be filtered and provided as input for Base Quality Score Recalibration |
| Command | java -jar -T HaplotypeCaller -R ref -I realigned_reads.bam -o raw_variants.vcf |
| Step 9 | Extract SNPs & Indels |
| Tool | GATK |
| Input | raw_variants.vcf, reference genome |
| Output | raw_indels.vcf, raw_snps.vcf |
| Notes | This step separates SNPs and Indels so they can be processed and used independently |
| Command | java -jar -T SelectVariants -R ref -V raw_variants.vcf -selectType SNP -o raw_snps.vcfjava -jar GenomeAnalysisTK.jar -T SelectVariants -R ref -V raw_variants.vcf -selectType INDEL -o raw_indels.vcf |
| Step 10 | Filter SNPs |
| Tool | GATK |
| Input | raw_snps.vcf, reference genome |
| Output | filtered_snps.vcf |
| Notes |
The filtering criteria for SNPs are as follows: QD < 2.0 Note: SNPs which are ‘filtered out’ at this step will remain in the filtered_snps.vcf file, however they will be marked as ‘basic_snp_filter’, while SNPs which passed the filter will be marked as ‘PASS’ |
| Command | java -jar -T VariantFiltration -R ref -V raw_snps.vcf --filterExpression 'QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0 || SOR > 4.0' --filterName "basic_snp_filter" -o filtered_snps.vcf |
| Step 11 | Filter Indels |
| Tool | GATK |
| Input | raw_indels.vcf, reference genome |
| Output | filtered_indels.vcf |
| Notes |
The filtering criteria for SNPs are as follows: QD < 2.0 Note: Indelss which are ‘filtered out’ at this step will remain in the filtered_indels.vcf file, however they will be marked as ‘basic_indel_filter’, while Indels which passed the filter will be marked as ‘PASS’ |
| Command | java -jar GenomeAnalysisTK.jar -T VariantFiltration -R ref -V raw_indels.vcf --filterExpression 'QD < 2.0 || FS > 200.0 || ReadPosRankSum < -20.0 || SOR > 10.0' --filterName "basic_indel_filter" -o filtered_indels.vcf |
| Step 12 | Base Quality Score Recalibration (BQSR) #1 |
| Tool | GATK |
| Input | realigned_reads.bam, filtered_snps.vcf, filtered_indels.vcf, reference genome |
| Output |
recal_data.table* *Intermediary file, removed from final output |
| Notes | BQSR is performed twice. The second pass is optional, but is required to produce a recalibration report. |
| Command | java -jar GenomeAnalysisTK.jar -T BaseRecalibrator -R ref -I realigned_reads.bam -knownSites filtered_snps.vcf -knownSites filtered_indels.vcf -o recal_data.table |
| Step 13 | Base Quality Score Recalibration (BQSR) #2 |
| Tool | GATK |
| Input | recal_data.table, realigned_reads.bam, filtered_snps.vcf, filtered_indels.vcf, reference genome |
| Output |
post_recal_data.table *Intermediary file, removed from final output |
| Notes | The second time BQSR is run, it takes the output from the first run (recal_data.table) as input |
| Command | java -jar GenomeAnalysisTK.jar -T BaseRecalibrator -R ref -I realigned_reads.bam -knownSites filtered_snps.vcf -knownSites filtered_indels.vcf -BQSR recal_data.table -o post_recal_data.table |
| Step 14 | Analyze Covariates |
| Tool | GATK |
| Input | recal_data.table, post_recal_data.table, reference genome |
| Output | recalibration_plots.pdf |
| Notes | This step produces a recalibration report based on the output from the two BQSR runs |
| Command | java -jar -T AnalyzeCovariates -R ref -before recal_data.table -after post_recal_data.table -plots recalibration_plots.pdf |
| Step 15 | Apply BQSR |
| Tool | GATK |
| Input | recal_data.table, realigned_reads.bam, reference genome |
| Output | recal_reads.bam |
| Notes | This step applies the recalibration computed in the first BQSR step to the bam file. |
| Command | java -jar -T PrintReads -R ref -I realigned_reads.bam -BQSR recal_data.table -o recal_reads.bam |
| Step 16 | Call Variants |
| Tool | GATK |
| Input | recal_reads.bam, reference genome |
| Output | raw_variants_recal.vcf |
| Notes | Second round of variant calling performed on recalibrated bam |
| Command | java -jar GenomeAnalysisTK.jar -T HaplotypeCaller -R ref -I recal_reads.bam -o raw_variants_recal.vcf |
| Step 17 | Extract SNPs & Indels |
| Tool | GATK |
| Input | raw_variants_recal.vcf, reference genome |
| Output | raw_indels_recal.vcf, raw_snps_recal.vcf |
| Notes | This step separates SNPs and Indels so they can be processed and analyzed independently |
| Command | java -jar GenomeAnalysisTK.jar -T SelectVariants -R ref -V raw_variants_recal.vcf -selectType SNP -o raw_snps_recal.vcfjava -jar GenomeAnalysisTK.jar -T SelectVariants -R ref -V raw_variants_recal.vcf -selectType INDEL -o raw_indels_recal.vcf |
| Step 18 | Filter SNPs |
| Tool | GATK |
| Input | raw_snps_recal.vcf, reference genome |
| Output | filtered_snps_final.vcf |
| Notes |
The filtering criteria for SNPs are as follows: QD < 2.0 Note: SNPs which are ‘filtered out’ at this step will remain in the filtered_snps_final.vcf file, however they will be marked as ‘basic_snp_filter’, while SNPs which passed the filter will be marked as ‘PASS’ |
| Command | java -jar GenomeAnalysisTK.jar -T VariantFiltration -R ref -V raw_snps_recal.vcf --filterExpression 'QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0 || SOR > 4.0' --filterName "basic_snp_filter" -o filtered_snps_final.vcf |
| Step 19 | Filter Indels |
| Tool | GATK |
| Input | raw_indels_recal.vcf, reference genome |
| Output | filtered_indels_final.vcf |
| Notes |
The filtering criteria for SNPs are as follows: QD < 2.0 Note: Indelss which are ‘filtered out’ at this step will remain in the filtered_indels_recal.vcf file, however they will be marked as ‘basic_indel_filter’, while Indels which passed the filter will be marked as ‘PASS’ |
| Command | java -jar GenomeAnalysisTK.jar -T VariantFiltration -R ref -V raw_indels_recal.vcf --filterExpression 'QD < 2.0 || FS > 200.0 || ReadPosRankSum < -20.0 || SOR > 10.0' --filterName "basic_indel_filter" -o filtered_indels_recal.vcf |
| Step 20 | Annotate SNPs and Predict Effects |
| Tool | SnpEff |
| Input | filtered_snps_final.vcf |
| Output | filtered_snps_final.ann.vcf, snpeff_summary.html, snpEff_genes.txt |
| Command | java -jar snpEff.jar -v snpeff_db filtered_snps_final.vcf > filtered_snps_final.ann.vcf |
| Step 21 | Compute Coverage Statistics |
| Tool | Bedtools |
| Input | recal_reads.bam |
| Output | genomecov.bedgraph |
| Notes | Load the genomecov.bedgraph file into IGV to view a coverage map at the entire genome or chromosome level |
| Command | bedtools genomecov -bga -ibam recal_reads.bam > genomecov.bedgraph |
| Step 22 | Compile Statistics |
| Tool | parse_metrics.sh (in house) |
| Input | alignment_metrics.txt, insert_metrics.txt, raw_snps.vcf, filtered_snps.vcf, raw_snps_recal.vcf, filtered_snps_final.vcf, depth_out.txt |
| Output | report.csv |
| Notes | A single report file is generated with summary statistics for all libraries processed containing the following pieces of information:
|
This pipeline was presented at the NYU-HiTS seminar on March 03, 2016. Slides available here.


23 Comments
Minju Kim · 2016-11-29 at 2:12 pm
Hi,
I am Minju, a master student of Dept. of Biology, performing computational research with HTS data.
my question is that where could I get the file, GenomeAnalysisTK.jar. because the Broad Institute, the developer, only provide GenomeAnalysisTK.tar which means I can’t execute the commands(specifically I am stuck in the Step 8: raw variant calling)
“Error: Unable to access jarfile GenomeAnalysisTK.jar”
error message from the mercer.
I want to ask you some solutions either for A or B.
A. how can I modify the command given in Step 8, if I want to call the variants with GATK.tar
B. where should I get GATK.jar
Mohammed Khalfan · 2016-11-29 at 4:07 pm
Hi Minju,
On mercer, you need to call the full path to the GATK jar file (after loading the GATK module). Your command should look like this:
java -jar /share/apps/gatk/3.3-0/GenomeAnalysisTK.jar -T HaplotypeCaller -R ref -I realigned_reads.bam -o raw_variants.vcf
Nadia · 2019-09-25 at 8:54 am
.tar is the compressed form of the GATK tool.
uncompressed it using a linux command
tar -zxvf yourfile.tar.gz
Wilfrid Carre · 2017-01-12 at 6:21 am
Hi,
I use more or leass the same tools.
Several comments on your pipeline:
Where do you put the read groups in the BAM that are necessary for GATK ?
The Base Quality Score Recalibration from GATK works with dbSNP and not with the variant you just call for the -knownSites option. Read the manual from GATK : https://software.broadinstitute.org/gatk/gatkdocs/org_broadinstitute_gatk_tools_walkers_bqsr_BaseRecalibrator.php
The point of the recalibration is to use known genuine SNP in order to improve your calling.
Mohammed Khalfan · 2017-01-12 at 7:22 am
Hi Wilfrid,
Thanks for your feedback. Answers to your questions:
1) The read groups are provided during the alignment step. I’ve updated the post to include this (step 1)
2) You are correct, and where possible you should use a ‘gold standard’ set of variants for the BQSR step. Unfortunately, this data is not available for all organisms, and in these cases, it is necessary to do what is known as ‘bootstrapping’ to produce a set of variants to use at this step, which is what I have demonstrated above. See: http://gatkforums.broadinstitute.org/wdl/discussion/1247/what-should-i-use-as-known-variants-sites-for-running-tool-x
See “Important Notes” above: The pipeline assumes no known variants are available for the Base Quality Score Recalibration step and as such bootsraps a set of SNPs and Indels to provide as input at this step.
Kai Feng · 2017-01-14 at 10:54 am
Hi there,
your pipeline is quite straightforward and promising, I figured out this was done on only one sample, can you provide a detailed instruction on multiple samples?
Thanks,
Kai
Malisa · 2017-02-19 at 11:43 am
Hi,
I am a PHD student and trying to run the codes step by step. When I followed the steps and ran Step 10, there were warnings said “undefined variable MQRankSum”. How can I solve this problem? Thanks for your time.
Mohammed Khalfan · 2017-02-22 at 11:41 am
Hi Malisa,
Is this happening only for certain lines? If so, I believe this means that there is no MQRankSum annotation for that particular site. In this case, you can ignore these warnings.
David willmore · 2017-02-21 at 10:04 am
Hi Mohammed,
The pipeline you elaborated is good, please keep up the good work. It will help many. You have explained it well and kept it simple and lucid.
well, i was wondering, from where “depth_out.txt” you are sourcing out?
just i am curious to know…
Keep up the good work !!!
Cheers,
David
Mohammed Khalfan · 2017-02-22 at 11:32 am
Hi David,
Thanks for the kind words! the ‘depth_out.txt’ file is created using the samtools depth utility, see Step 3 above (Collect Alignment & Insert Size Metrics).
irene · 2017-02-23 at 11:20 am
Hi David really nice work!
Just a quick question. The GATK website says that you actually don’t need to run IndelRealigner if you are going to use Haplotypecaller. But this is in your steps. Would you recommend it anyway?
thanks
ib
Mohammed Khalfan · 2017-03-12 at 2:56 pm
Hi Irene, thanks for the feedback! At the time of writing this post, IndelRealigner was a recommended step in the GATK best practices. With the latest version of Haplotypecaller, it seems this step is no longer required, so I think you are safe to skip it.
irene · 2017-02-23 at 11:21 am
Apologies misread your name Mohammed 🙂
Reza · 2017-03-09 at 3:46 pm
Hi
In step 10 i want to add “GQ < 30" but GQ is not in INFO field, how can i do it? it must be as following order.
–filterExpression "QUAL 60.0 || MQ < 30.0 || DP < 10 || GQ < 30"
Thanks in advance
Gepoliano Chaves · 2017-03-30 at 10:18 am
Hi Mohammed,
Thanks for your pipeline, it helped me a lot on calling SNPs from the files I work with. Now, I have a question about statistics, how can I know that the SNPs I have identified in this pipeline are actually significant? Which statistical test should I choose, if so, and how do I apply such test to the data-set?
Deepa Krishna · 2017-05-25 at 10:20 am
Hi,
I am a Masters student, I was looking for something similar to workflow presented here and am glad I came across your work.
You mentioned about Compile Statistics in step 22, a Tool, parse_metrics.sh (in-house). Am interest in knowing how it works and I would like to use it to produce the report.
Thank you
Bandana · 2017-06-02 at 12:12 pm
I have one question, can I apply this pipleline for plant viruses? Also, how can I create a custom SnpEff Database?
Tom · 2017-09-22 at 11:25 am
Hi
I’m wondering if an extra “SelectVariants” step is necessary to select only SNP with “PASS” tag after step 10 before used as knownsites in Step12. I’ve seen an example doing SelectVariants with -select ‘vc.isNotFiltered()’. Does BaseRecalibrator use SNP with ‘PASS” tag only in filtered_snps.vcf? I appreciate your insights.
Mohammed Khalfan · 2017-09-28 at 3:29 pm
Hi Tom,
This is not necessary. BQSR will only consider SNPs with “PASS” when doing recalibration, so you can give it filtered_snps.vcf directly.
nord · 2017-10-13 at 3:07 am
your pipeline is good. Can you recommend your paper using this pipeline to analyse to me? Thanks very much.
irene · 2017-11-19 at 9:15 am
Hi Mohammed,
can you run step 20 if you have done steps 16-19 in g mode?
if yes, can you please share the command?
thanks,
ib
D. Bautista PhD · 2018-01-14 at 11:39 pm
Your pipeline is quite thorough. I’m wondering if this is available to non NYU users.
Thippesh Sannasiddappa · 2018-06-20 at 7:14 am
Hi Mohammed,
Thanks for posting nice and detailed analysis pipeline for variant calling. I am doing bacterial genome SNP/indel calling. I have few questions to ask;
1. Which versions of GATK, SnpEff, Samtools, Bedtools were used in your pipeline?
2. What parameters you suggest for filtering SNPs/Indels for bacterial genomes?
3. What these parameters mean: QD 200.0, ReadPosRankSum 10.0? Are there any cut off standards for calling SNPs/Indels for bacterial genomes?